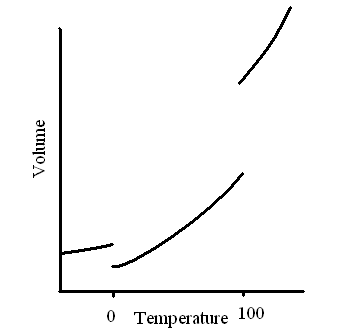
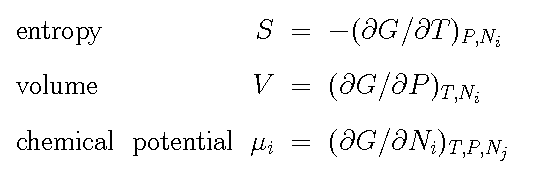The lines in the T-P diagram are ZPF lines but there are
two phases with zero amount along the lines! As T and
P are intensive variables the change from one phase to
another across a line is immediate but what is not shown in this
diagram is that crossing a line means a change in volume and requires
or releases an amount of heat. We know that it requires heat to melt
ice and a mixture of ice and water remains at the same temperaure, 0
oC (at atmospheric pressure) until all ice has melted.
Question: Why are there always 3 lines meeting at each invariant point
in the Fe-TP phase diagram?
Select answer:
- a) because there are 3 phases stable at the point and
one of these phases is stable along each line.
- b) because there are 3 phases stable at the point and
each line represent the exclusion of two of these.
- c) because there are 3 phases stable at the point and
each line represent a combination of two of these.
hint-ab: according to Gibbs phase rule the lines must have 2
stable phases
correct-answer-c
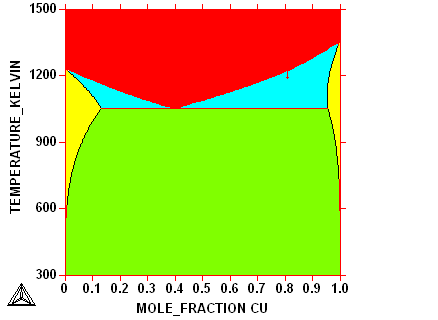
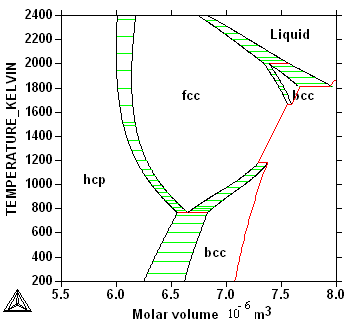
Question: Why is there a gap between the two two-phase regions fcc+bcc?
Question: Why are the tie-lines horizontal?
Phases with a high entropy become
more stable with increasing temperature. In general less ordered and
less dense phases, for example the liquid, have higher entropy, the
phonon frequecies are higher?? However, entropy also depend on
temperature and its value can change drastically for example with a
second order transion like ferro-magnetism in iron. The Gibbs energy
for a phase must always decrease with increasing temperature because
entropy must always be positive.
Phases with large molar volumes decrese
their stability more than close packed dense phases with increasing
pressure. The Gibbs energy for a phase increase with increasing
pressure because volume must always be positive.
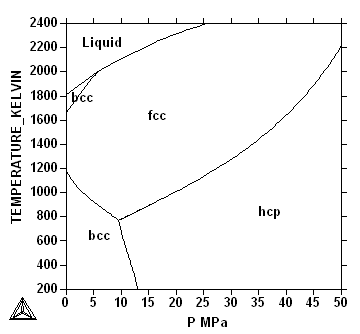
In Fig.~\ref{fg:fe-sp} the phase diagram for
pure Fe is plotted with S and P as axis. As
S is an extensive property the values of the entropy are
different in the two phases in equilibrium and the sinlge line in the
T-P phase diagram becomes two lines and in between one can
draw tie-lines connecting the points of equilibrium between the two
phases.
Question: Why are the tie-lines vertical?
The term fluid is used to indicate the aqueous
phase at high pressure and temperature where the gas and liquid are
treated as the same phase. This means one must use the Helmholtz
energy for the modeling.
The word eutectic is greek and means
originally ``fluent'' (lättflytande) and has been used to indicate the
equilibrium where the liquid is stable ?????
The congruent point represent an
equilibrium between two phases in a binary or higher order system
where both phases have the same composition.
The maximum of a miscibility gap is called
a consolute point.
In Fig.~\ref{fg:fe-sv} the phase diagram for
pure Fe is plotted with S and V as axis. Now both
axis quantities are extensive and the tie-lines in the two-phase
regions between the phases that are in equilibrium can have an
arbitrary slope.
Question: Why are the tie-lines not parallel to the axis?
Question: There some special cases when one can predict the
slope of the tie-lines, which?
a) at a congruent transformation
Explanation of H2O PD with critical
point for the gas/liquid equilibrium line and the backward sloping
equilibrium between liquid/ice.
Question: What is the important physical effect of the density of
ice lower than liquid water?
a) ice floats on top of water
Question: Another less important effect?
a) glaciers can pass obstacles by meling on the high pressure side
and freeze on the low pressure side.
A phase with a miscibility gap can exist
simultaneously with (at least) two different sets of some extensive
variables, for example volume or composition. One example is the
H2O system for which one can pass from the gas to the liquid
state at high pressure and temperature. This means that above
a certain limit of temperature and pressure gas and liquid is
the same phase, usually called fluid.
Unless you work with fluids, either in geology or in steam
generators, you will much more frequenly come across miscibility gaps
where the phase can exist at the same time with (at least) two
different compositions. Such miscibility gaps exists in the liquid
and solid phases and they may often be metastable because other phases
are more stable.
Question: Can one have a phase with a miscibility gap in S?
Describe the invented unary with S1, S2, liquid and gas
Short about thermodynamic models, EOS for pure elements
There is a kind of diagram frequently
used on its own or as complement to phase diagrams in thermodynamics
that is called "property diagram".
Property diagrams for the different phases of Fe
G and V and S and Cp curves at constant P and varying T
G and V and S and Cp curves at constant T and varying P
Cp and Cv curves and their relation
Properties of the invented unary.
Show how a change of Cp for a phase affects the phase diagram (phase
comming back at high T)
Show the risk of negative S or Cp when modeling
Question: Why is negative Cp impossible in a real phase?
CARNOT CYCLES
The steam engine and the refrigerator
The phase diagram you will find most often are the binary phase
diagram with the composition on the x axis and temperature on the y
axis and for constant pressure, usually at 1 bar. These diagrams are
so common that some people think it is the only kind of phase diagram.
But this tutorial have already shown you a lot of other kinds of phase
diagrams and you will at the end of the tutorial you will find these
diagrams almost trivial considering how many other kinds of
interesting phase diagrams one can use.
The step from unary phase diagram to binary means one has one more
component in the system. As already stated the size of the system has
no influence on the equilibrium and normally one uses mole fractions,
mass (weight) fractions or percent to give the composition. The
composition is an extensive variable, the conjugate state variable is
the chemical potential of the component. Phase diagrams with a
chemical potential axis instead of composition axis are very similar
to a unary phase diagram, the regions are single phase and along the
line there are two phases stable and where the lines meet there are
invariant 3 phase equilibria, see Fig~\ref{fg:agcuat}. The same phase
diagram plotted with a composition axis is shown in
Fig.~\ref{fg:agcuxt}. Changing from chemical potential to composition
is thus similar to change from pressure to volume in unary systems.
We have horizontal tie-lines connecting the phases in
equilibrium. c
But we are advancing a bit too fast, first let us go back and
consider what it means to have two components mixing in the same
phase.
In a gas phase the molecules can move
freely and if the gas is ideal they are non-interactive and all
collosions are elastic without loss of kinetic energy. In statistical
mechanics one can derive the increase of entropy mixing two monoatomic
gases of elements A and B and the following formula is found
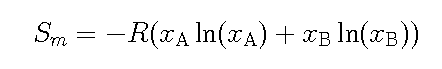
where xi is the mole fraction of the element.
Much time can be spent on explaining how different elements can be
formed and one can formulate several chemical reactions between the
elements but in the section on thermodynamics the Gibbs energy for an
ideal gas with several molecules i is derived
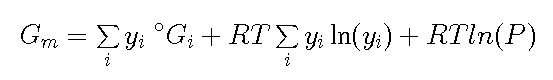
where yi is the fraction of molecule i
and oGi is the Gibbs energy of
formation of the molecule from the elements in their standard
state.
From high school chemistry you may be familiar with writing
chemical reactions between molecules and using equilibrium constants
to determine their fractions but in computational thermodynamics with
many components and molecules it is more convenient to describe the
Gibbs energy of a phase with equations like this and the equilibrium
is the same and the equilibrium constant can be calculated from the
relevant oGi.
Most of the phase diagrams we will deal with here will not include
the gas phase, only liquid and solid phases. Non-ideal gases is not
considered at all but can of course be modeled like the condenced
phases although traditionally other types of models are used for
fluids.
In the liquid phase that atoms are much closer than in the gas and
there are attractive or repulsive forces between the atoms. If the
atoms are interacting weakly one will have the same entropy of mixing
as in the gas and the most used model for a metallic liquid phase
is
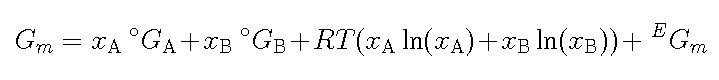
where EGm describes the non-ideal
part of the mixing. All these models and equations are described in
more detail in the thermodynamics course
There can be many solid phases in a binary system and this will be
discussed more later. All stable solid phases are crystalline
i.e. the atoms are arranged in fixed lattices. Most of these are very
simple like bcc, hcp and fcc shown in Fig.~\ref{fg:lattice1}. Only in
special cases, like rapid quenching, can one obtain an amorpheous
solid phase where the atoms are arranged randomly on space similarly
to the liquid but contrary to the liquid the atoms cannot move
around.
Even in crystalline solids the atoms can arrange themselves
randomly on the available sites and simple statistics shows that
one will have an entropy of mixing identical to that for an
ideal gas. The Gibbs energy expression for a crystalline solid
phase with substitutional mixing on the lattice is
This is identical to the model for the liquid! So there is no real
difference between a substitutional model for crystalline phases and
for the metallic liquid phase. We will later find cases when the
lattice have several different types of sites and where the atoms does
not mix randomly but for many simple cases the equations above are
sufficient for modeling the thermodynamic properties of a phase.
To denote different solid phases in binary
phase diagrams one has traditionally used greek letters like \alpha,
\beta, \gamma starting from the low temperature phase to the left.
This labelling is useless and confusing when extending to higher order
systems so in this tuturial the crystalline phases will be named after
their "Strukturberich", a method based on the crystalline structure.
For the cases when a phase have no Strukturbericht we will use the
"prototype" as name of the phase. This is furher elaborated in the
thermodynamics course.
There is an important method to determine the
amounts of the phases along a tie-line called the lever rule.
Different points on the tie-line represent different overall
compositions but as the phases in equilibrium have fixed compositions,
given by the end points of the tie-line, the system can only change
its overall composition by changing the amounts of the phases. At one
end of the tie-line one have zero amount of the other phase and along
the tie-line that amount increases to reach 100\% at the other end.
One can illustrate this put using a "lever" with the balancing
point at the overall composition and the amounts of the phases as
weights at each end. The long arm will have a low amount and the
short a high amount as shown in Fig.~\ref{fg:leverule}
The solubility curves for liquid and
solids have tradionally been given special names, the one giving the
solid phase in equilibrium with liquid is called "solidus" and the one
giving the liquid in equilibrium with solid is called "liquidus".
An alloy with a given composition will thus on heating start
melting at its liquidus temperature and on cooling from a liquid state
it will start solidifying at the liquidus temperature.
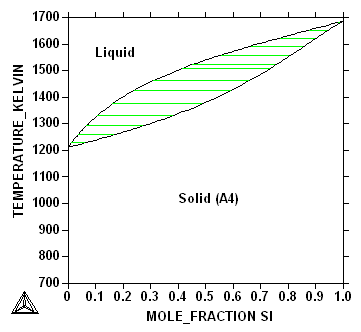
At the melting point for Ge in Fig.~\ref{fg:sige} a small addition
of of Si will increase the melting point. For the pure elements the
transformation from liquid to solid occurs at a singe temperature but
for an alloy with both elements there will be a solidification
interval where the liquid and solid will coexist over a temperature
range, called the solidification interval, see Fig.~\ref{fg:gesi2}.
At the Si side the solubility curves join each other at the melting
point for pure Si.
In some applications, for example soldering, one is interested in
as small solidification interval as possible. In casting of metals it
is often convenient to have as small solidification interval as
possible to avoid shrinkage and pores in the cast. See also the
section on segregation later.
It is not obvious that an addition of an element with a higher
melting temparure will increase the melting temperature. In
Fig.~\ref{fg:crfe} for the Cr-Fe system show that one may have more
complicated behaviour with a minimum (or maximum in other cases) in
the liquidus curves.
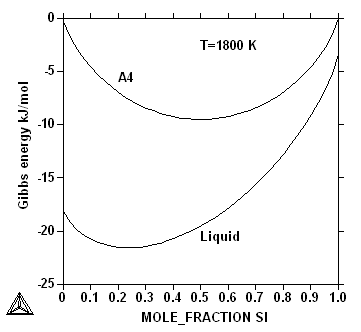
The Gibbs energy curves for the liquid and the diamond (A4) phase
at different temperatures are shown in the figures below.
All ZPF lines in all phase diagrams represent such common tangent
constructions. In ternary and higher order systems there will be
tangent planes or hyper-planes, where the Gibbs energy curves of the
co-existing phases have a plane in common. This is a consequence of
the fact that the chemical potentials of all elements must be the same
in the phases that are in equilibrium. When modelling phase diagram
this can be used as information to determine the model parameters of
each phase.
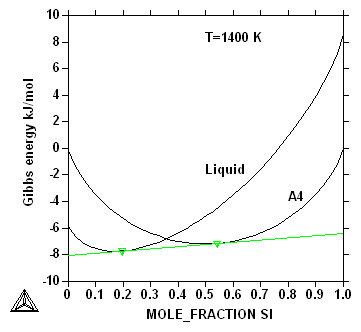
The point of crossing between the Gibbs energy
curves is not without importance. When an alloy is rapidly quenched
equilibrium cannot be established and the crossing point marks the
composition for which the alloy can be transformed from liquid to
solid (A4) without any diffusion. For solid state transformations
such "T-zero" or T0 points can be important to control
diffusionless transformation like martensite in steels.
We will now look at the connection between the phase diagram
and the microstructure for a simple eutectic phase diagram. We will
assume that the solid phases will form dendrites and there will
then be a cooperative eutectic growth. In the figures below
you will be able to see simultaneously how the composition of the
phases in the phase diagram and the microstucture changes during
the cooling. In the 3rd frame you can select yourself a
quantity to plot.
First select composition
| phase diagram |
microstructure |
| user selected: DTA curve |
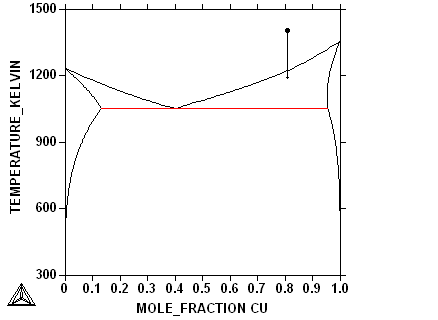 |
 |
 |
| third question |
answer |
\endslide
Eutectic phase diagrams. Microstructure at various compositions
Peritectic phase diagrams. Microstructure at various compositions
T-x and T-mu diagram
show G-x diagrams for variable T and corresponding T-x (movie?)
allow student to enter model paranmeters and calculate G-x
and T-x diagrams
Other special invariants (monotectic, congruent melting ....??)
Liquid miscibility gap
Solid miscibility gap, metastable miscibility gaps
Modeling binary phases. Lattice stabilities, heat capacity of
intermetallic phases (Kopp-Neuman). Gibbs energy of mixing.
Examples of calculated diagrams with different parameters.
Phases with restricted solubility
Intermetallic phases
Phase diagrams with several features
Chemical ordering
Solid state transformations, eutectoid, peritectoid etc
Carbides, oxides etc
Metastable phase diagrams
Scheil-Gulliver solidification simulation
Variable pressure
Quasi-binary phase diagrams
Isothermal section
Projections and sections
Ternary Isopleth diagrams (Al-Mg2Si)
Solubilities of third element in binary phases
Ternary phases
Ternary diagrams with fix activity
3D phase diagrams
Gibbs energy surfaces
Para-equilibrium phase diagrams
Phase diagrams for geological systems including high pressure
Isopleths
Property diagrams
Congratulations! You have finished the tutorial and your score is
very good ???? . You have the now the necessary background to use
phase diagrams in your future studies or other work.
Not in any particular order